病因
病因
病因:
1.分型 根据形态学和血清学特征,将CDA分为以下3种类型:
Ⅰ型:巨红细胞伴骨髓巨幼样改变和核间染色质桥。
Ⅱ型:正红细胞或巨红细胞伴骨髓双核红、多核红、核碎裂等,酸溶血试验阳性。
Ⅲ型:巨红细胞伴骨髓12个核以上的多核红(巨型有核红细胞)。
CDA Ⅰ型和Ⅲ型主要是靠形态学特征诊断的,CDAⅡ型又称作HAMPAS,是遗传性多核幼红细胞伴酸溶血试验阳性(hereditary erythroblastic multinuclearity with a positive acidified serum test,HAMPAS)的简称。另外文献报道的50多例患者,其疾病特征不符合上述分型。
2.病因 CDA是一种常染色体遗传性疾病,但尚不清楚,体外培养红细胞集落生成正常,但可见多核红细胞,可能由于红系核膜和胞浆膜的结构异常所致,可见幼红细胞破坏增加和DNA减少。
(1)CDAⅠ型:为常染色体隐性遗传性疾病,已报道了100多例,其中部分患者存在血缘关系,呈家族性发病。起病年龄从出生到成年,平均年龄10岁,男女比例1.1∶1。
(2)CDAⅡ型(HEMPAS):是CDA中最多见的一种类型,已有200多例报道,部分患者有血缘关系,是一种常染色体隐性遗传性疾病。发病年龄从出生到成人,平均年龄14岁,男女比例为0.9∶1。
(3)CDAⅢ型:是一种少见的类型,至今报道70余例,主要来自大约16个家族。男女比例为0.8∶1。诊断时的平均年龄是24岁,从出生到老年均可发病。
(4)其他类型CDA:CDA各型鉴别困难,尚有50多例患者不能被划分到以上3型,因此提出了变异型的概念。文献报道中,CDAⅣ型的特点为骨髓形态类似于CDAⅡ型,10%~40%的有核红细胞为双核。酸溶血试验阴性,有1例家庭为常染色体显性遗传,与CDAⅡ型不同。红细胞表面i抗原不增加。大部分患者贫血症状较轻,临床预后相对较好。
发病机制
发病机制:患者铁代谢加快,是正常人的10倍,骨髓中幼红细胞增生旺盛,致肠道对铁的吸收增加,运铁蛋白饱和度增高,而铁利用率下降,铁清除增加。骨髓中幼红细胞破坏增加,仅30%的红细胞能进入外周血。这些进一步证明了红系无效造血。患者间接胆红素和乳酸脱氢酶增高,结合珠蛋白水平降低,转铁蛋白饱和度增加。红细胞寿命轻度缩短。部分患者HbA2增高,珠蛋白链合成不平衡,非-α/α链比例降低(0.5~0.7)。酸溶血试验阴性,红细胞i抗原效价正常。
CDAⅠ型的缺陷在干细胞水平,但细胞培养CFU-E和BFU-E的量正常,是因为骨髓中存在正常和异常细胞的混合克隆,提示每一个干细胞的子代中异常的表达具有多样性。CDAⅠ型的基因(CDAN1)定位于15q15.1~15.3,但有些患者未发现这种基因定位,提示遗传的异质性。
临床表现
临床表现:发病年龄和贫血轻重差别极大,常在10岁以后才得到确诊。起病缓慢,多因贫血而就诊,并间断地出现
黄疸和尿色的改变,肝、
脾肿大,可见胆管梗阻现象。病程久的可继发含铁血黄素沉着症和血色病。含铁血黄素沉着虽与多次输血有关,但经实验证明主要由于铁代谢异常,骨髓红细胞系增生旺盛而导致肠道对铁的吸收增加。运铁蛋白饱和度增高,
59Fe利用率下降,清除增快。有些病人并有红细胞寿命缩短。成人患者中可见性腺发育障碍、甲状腺功能低下和非家族性
糖尿病等表现,个别病人可继发
肝硬化。
贫血轻重不等,可见非特异性的变形红细胞,网织红细胞与贫血成比例地降低。白细胞和血小板正常。血清未结合胆红素增高,尿胆原常呈阳性。骨髓中可见胞体大的多核幼红细胞。
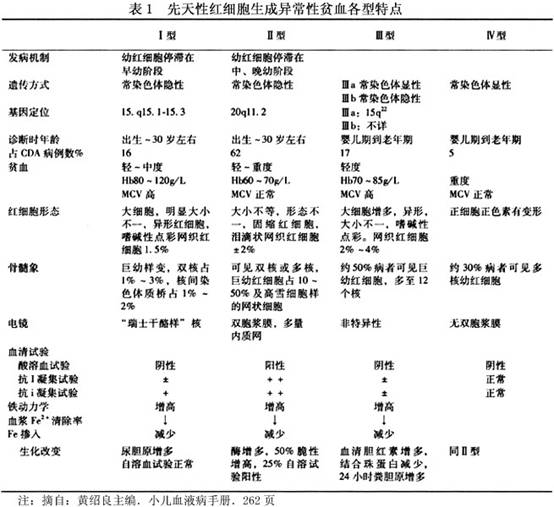
根据血象、骨髓象和生化等方面的改变,将此症分为4型,各型的特点见表1。
最常见的是Ⅱ型,约占本症的60%,又称为遗传性多核幼红细胞伴阳性酸溶血试验(hereditary erythroblastic multinuclearity with a positive acidified serum,HEMPAS)。常见贫血与轻度
黄疸,肝、
脾肿大。约半数红细胞脆性增高,红细胞形态不规则,嗜多色性,偶见有核红细胞。骨髓中幼红细胞增多,可见双核及多核幼红细胞。电镜检查可见有的幼红细胞有双层胞浆膜。推测此种变化是细胞不易分裂的原因;尚可见戈谢样细胞。酸溶血试验阳性,在抗i和抗I血清中溶血增加。曾有报道此型的红细胞膜蛋白的糖基化作用异常。北京儿童医院于1979年曾见到1例5岁男孩,具有上述的典型症状及化验检查结果,骨髓中可见较多的多核中、晚幼红细胞(图1)。
治疗
治疗:多数病人不依赖输血也可维持健康水平。重症病人则需反复输血,或做脾切除以减少输血次数,但需注意切脾后发生严重感染。合并胆结石的需做胆囊切除。禁忌服用铁剂。应用维生素B
12、B
6、
叶酸和维生素E无效。对于少数依赖输血而致含铁血黄素沉着的病人,目前采取放血和铁螯合剂治疗,可采用
去铁胺皮下注射。
研究发现在体外,EB病毒转化的CDAⅠ型患者的B淋巴细胞产生干扰素α水平明显少于正常细胞,提示CDAⅠ型患者产生或释放干扰素α障碍是此病潜在的发病机制。应用干扰素α治疗取得了疗效。
已有同胞间成功骨髓移植的报道。
预后
预后:
1.CDAⅠ型 20%的患者需输血治疗。常用的补血治疗和皮质激素治疗无效,虽然
脾大常见,部分患者进行了脾切除手术,但贫血的症状无改善。一些患者发生胆结石,需行胆囊切除术。
由于肠道吸收铁增多,红系无效造血和轻微溶血引起的含铁血黄素沉着症是严重的远期并发症。已有同胞间成功骨髓移植的报道。
2.CDAⅡ型(HEMPAS) 贫血严重的患者需要输血治疗。70%的CDAⅡ型患者脾切除治疗有效,但脾切除术不能预防铁的进一步蓄积。由于输血和胃肠道吸收铁增加,甚至一些未输血的患者,也可出现铁负荷过重。
肝硬化和心脏并发症是主要的死亡原因。放血和去铁胺治疗有效。多数患者出现胆结石,部分需行胆囊切除术。
3.CDAⅢ型 CDAⅢ型患者很少需要输血或脾切除治疗。像其他类型的CDA一样,主要的合并症是血色病。部分患者可发生单克隆丙种球蛋白病、骨髓瘤、眼血管样条纹增多。